使用期限*
许可形式单机
原产地美国
介质下载
适用平台windows,mac
科学软件网是一个以引进国研软件,提供软件服务的营业网站,网站由北京天演融智软件有限公司创办,旨在为国内高校、科研院所和以研发为主的企业事业单位提供的科研软件及相关软件服务。截止目前,科学软件网已获得数百家国际软件公司正式授权,代理销售科研软件达一千余种,软件涵盖领域包括经管,仿真,地球地理,生物化学,工程科学,排版及网络管理等。同时,还提供培训、课程(包含34款软件,66门课程)、实验室解决方案和项目咨询等服务。
Assemble by Name is particularly helpful if you do a lot of sequencing and if you have numerous samples that are done with a standard set of sequencing primers. Some other applications of Assemble by Name can include:

Trim to Reference eliminates the ends of sequences that extend beyond an assembled Reference sequence.

Trim Vector removes sequence-specific data contaminating the ends of your sequences.

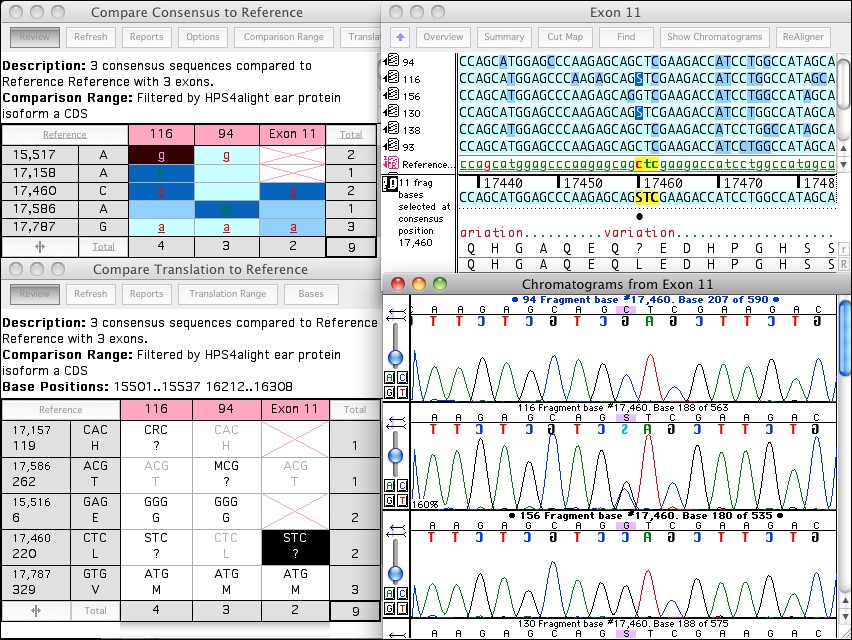
Sequencher DNA序列分析软件是科学家们*的工具。持续发展和改进**过25年,Sequencher提供了**的功能特征,成千上万的出版物都有相关的应用介绍。新手用户可以以少的时间投资产生结果,而有经验的用户会惊叹于功能的深度和控制。Sequencher自带各种专有算法,可以为Sanger, Next-Gen Sequencing (NGS)和RNA-Seq序列数据生成结果。
Maq
Maq是Sequencher v5.0时被添加到插件家族中的。流行的Maq算法将Next-Generation数据的单端和成对端对准参考序列。初设计为对齐Illumina-Solexa数据,可以对齐任何短的读取数据(63 bases或更少)。参考序列可能是FastA或GenBank的文件形式。Maq使用二进制格式压缩引用和读取文件。重要的是,Maq需要很少的内存来运行,这使得执行下一代测序更*。它也适用于大约二百万个阅读的小项目,尽管有可能把较大的项目拆掉,然后合并结果。您的对齐结果可以在Maqview或Tablet中查看。
初Maq是在命令行上运行的。Sequencher提供了一个简单、易于使用的接口,它可以保护您不受命令行的影响,也不必学习如何使用命令行参数。
SAMtools的变异调用
已经排序和对齐,接下来检查使用新的变体调用特征对变体进行对齐。您是否已经把您的读数与我们的参考引导对齐或者您已经将您的SAM/BAM文件导出到别处,您仍然可以使用SAMtools的变量调用检查变量。
此分析可用于SNP耐受性对齐、甲基化耐受性对齐或GSNAP中的新RNA A-I编辑耐受模式。然后将生成的VCF文件和SAM文件加载到您喜欢的浏览器中,如果您没有自己喜欢的浏览器,可以用Tablet—我们DNA-Seq工具分布的一部分。提供带有注释的GTF文件的Tablet,您还可以在特性中看到变体。
科学软件网不仅提供软件产品,更有多项附加服务免费提供,让您售后**!
http://turntech8843.b2b168.com